Toward informed batch correction for single-cell transcriptome integration
BCHSI Affiliated Faculty Peng He, PhD, assistant professor in the Department of Pathology, recently published in Nature Computational Science "Toward informed batch correction for single-cell transcriptome integration." He shares insights on this research:
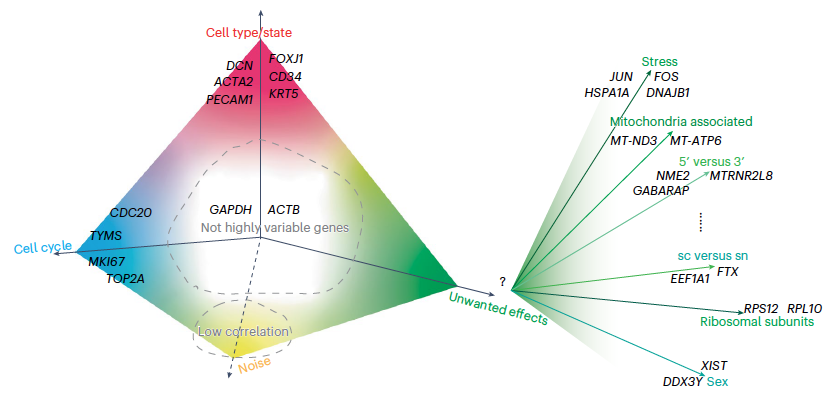
Modern technologies allow scientists to measure gene activity in thousands to millions of individual cells, helping us build detailed maps of tissues in health and disease. However, data generated in different labs or with different techniques often contain technical differences that make comparisons difficult. In this Perspective, we organize existing computational approaches into two broad categories — data cleaning methods that reduce known technical artifacts, and data integration methods that align datasets across experiments — and provide a clear, high-level overview of how these approaches work.
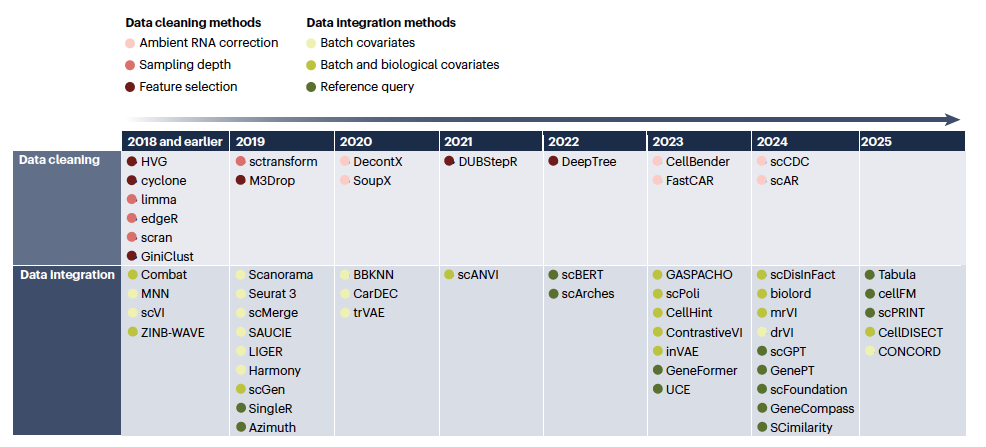
A central message of the paper is the need for greater interpretability. Many current models correct data effectively but offer limited insight into what was changed or why. We highlight key limitations in current practices — including inconsistent standards, limited transparency, and incomplete metadata — and advocate for more interpretable, knowledge-informed models. We also propose leveraging large, well-annotated cell atlases to train more robust and transparent integration frameworks, rather than repeatedly correcting small datasets in isolation.
Improving both robustness and interpretability is essential for building reliable reference maps and ensuring that discoveries from single-cell data are accurate and trustworthy. See full article here